
Vous n'êtes pas identifié.
• Annonce ToutSurTout
Déjà 15 ans !
Si vous souhaitez participer vous aussi aux discussions sur le forum, il faut vous inscrire ou vous identifier.
Les inscriptions sont actuellement OUVERTES.
Pages: 1
Réponse : 0 / Vues : 2 298
- Accueil forums
- » Le bar
- » Progeria (syndrome de Hutchinson-Gilford)
Message 1 Discussion postée le 25-02-2017 à 20:25:40
El Roslino 


- Titre: VIP
- Avancement: Niveau 5
- Lieu: U.S.A
- Date d'inscription: 07-07-2016
- Messages: 33 203
- Site web
Progeria (syndrome de Hutchinson-Gilford)
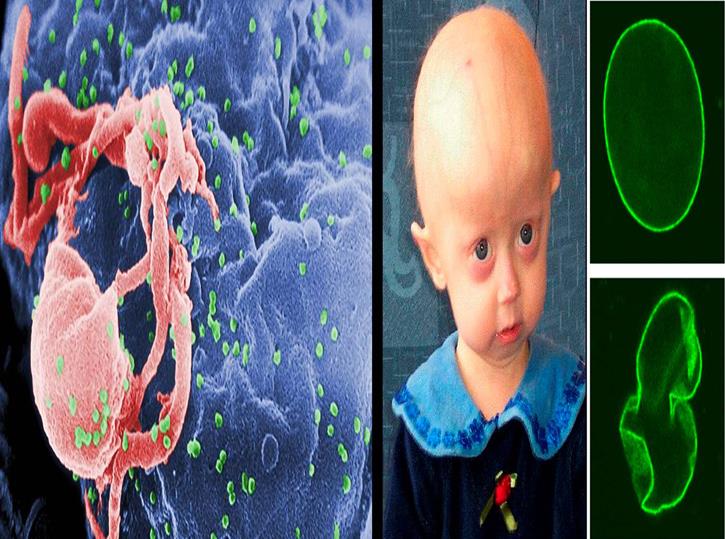
Le syndrome de Hutchinson-Gilford, plus communément appelé progeria, est une maladie génétique rarissime, affectant une naissance sur 4 à 8 millions, caractérisée par un vieillissement prématuré débutant dès la période néonatale. Elle est due à la mutation de novo (non présente chez les parents) d'un gène.
La maladie rarissime des "enfants-vieillards"
Progeria vient du grec "geron", le vieillard, et cette dénomination s'explique par les symptômes de la maladie : les enfants atteints souffrent d'alopécie (cheveux rares), ressentent des douleurs articulaires, ont une peau très fine et glabre, souffrent de troubles cardiovasculaires. Ils donnent l'impression d'un vieillissement accéléré, et leur stature ne connaît qu'une croissance lente. Leurs capacités cognitives ne sont en revanche nullement altérées. L'espérance de vie des patients atteints de progeria est actuellement très limitée : 12-13 ans en moyenne. Cette mortalité précoce est généralement causée par une athérosclérose ou un accident vasculaire cérébral. La progeria est une maladie rarissime : on ne connaît que 3 cas en France, 25 en Europe et une centaine dans le monde.
En 2003, l'origine de la progeria est identifiée
Longtemps, l'origine de la progeria a été une énigme : les hormones de croissance, vers lesquelles on dirigea d'abord les études, présentaient un niveau normal chez les patients. C'est en 2003 qu'une équipe française dirigée par Nicolas Lévy (unité Inserm UMR S 910 Génétique médicale et génomique fonctionnelle, faculté de médecine Timone - Université de la Méditerranée) a découvert la version mutée du gène impliquée dans plus de 90 % des cas connus de progeria. Appelé LMNA et situé sur le chromosome 1, le gène code normalement pour des protéines lamines A et C. Lorsque la mutation survient, ce gène produit une protéine tronquée, baptisée progérine, qui reste ancrée dans la membrane du noyau des cellules, s'y accumule, et entraîne finalement sa déformation et des dysfonctions.
Les laminopathies
La progeria s'inscrit dans la famille des laminopathies - ensemble de troubles liés à une altération des lamines A/C (deux protéines structurales de l'enveloppe nucléaire des cellules). Une dizaine de pathologies est déjà connue, ainsi que près de 200 mutations du gène LMNA : outre la progeria et les autres formes rares de vieillissement prématuré (syndromes de type lipodystrophie-Werner atypique, dysplasie acromandibulaire, dermopathie restrictive), les laminopathies incluent la dystrophie musculaire d'Emery-Dreifuss, la myopathie des ceintures de type 1B, la cardiomyopathie dilatée associée à des troubles de la conduction cardiaque, une forme spécifique de la maladie de Charcot-Marie-Tooth... Ces pathologies se caractérisent par un spectre large de troubles musculaires, cardiaques, neurologiques, osseux ou cutanés.
En 2008, un protocole d'essai clinique est conçu
En collaboration avec des chercheurs de l'université d'Oviedo, dirigés par Carlos Lopez-Otin, travaillant d'abord sur un modèle animal et un modèle cellulaire de la pathologie, Nicolas Lévy et Pierre Cau (Hôpital de la Timone, Inserm-APHM) sont partis à la recherche de molécules capables d'inhiber ou de bloquer la toxicité de la progérine. Ils ont montré qu'une association entre la pravastatine (statine, molécule utilisée pour prévenir les dépôts graisseux dans les vaisseaux) et le zolédronate (amino−bisphosphonate, médicament utilisé pour ralentir et corriger le processus d'ostéoporose) a des effets bénéfiques sur les cellules malades. L'espérance de vie de rongeurs présentant une anomalie génétique comparable à celle de la progeria a été considérablement allongée (179 jours contre 101). Ces résultats encourageants ont entraîné la mise au point d'un protocole de biothérapie, développé avec l'aide du Généthon et de l'AFM : d'une durée de 3 ans, il inclut 15 enfants européens atteints de la progeria. Si la guérison n'est pas encore en vue pour les jeunes malades, l'amélioration de leurs conditions de vie et l'augmentation de leur longévité semblent aujourd'hui deux objectifs réalistes.
Pour aller plus loin
Communiqués de presse
Progéria : résultats prometteurs d'une nouvelle thérapie génique chez l'animal26 octobre 2011)
Espoir pour la progeria, une maladie rare du vieillissement accéléré (13 juin 2008)(93,9 ko)
Des chercheurs français identifient le gène responsable d'un syndrome de vieillissement prématuré (Progeria) (16 avril 2003)(46,4 ko)
Inserm Actualités
Nicolas Lévy : Une lueur d'espoir dans la progéria (297,9 ko) - IA 210, juillet-août 2008, 25-29
Les associations de malades
Inserm-Associations - la base Inserm Associations
Sites
La progeria sur Orphanet (portail des maladies orphelines)
Institut de myologie : Focus sur les laminopathies (2005)
AFM : Espoir pour la progeria, une maladie rare du vieillissement accéléré (juin 2008)

Réponse : 0 / Vues : 2 298
Pages: 1
- Accueil forums
- » Le bar
- » Progeria (syndrome de Hutchinson-Gilford)
Le forum de Loic DL exécute les 7 requêtes SQL en 0.094 secondes.
© Copyright Numéro HFUF1C5 enregistré chez  pour toutsurtout.biz.
pour toutsurtout.biz.
Révision source V474
